RESEARCH
Precision Cancer Medicine
Why do some cancers come back after chemo/radiotherapy? From genetic points of view, cancer recurrence originates both at the cellular and organismal levels. At the cellular level, mutations in certain genes such as TP53 loss-of-function mutations dictate poor response to chemo/radiotherapy. At the organismal level, intra- and inter-tumoral clonal heterogeneity in the genome, epigenome, transcriptome and/or proteome renders the eradication of cancer cells by chemo/radiotherapy and immunotherapy more challenging. Precision cancer medicine aims to develop strategies: 1) to target cancer both at the cellular and organismal levels and 2) to prevent cancer before they acquire clonal heterogeneity.
Decoding a patient’s cancer genome is among the initial steps in devising a strategy to treat cancer. In clinical practice for solid tumors, the cancer genome information is primarily used to assess the eligibility of molecular targeted drugs. In contrast, in hematologic malignancies (HMs), the genome information also enables physicians to evaluate the patient’s prognosis and to determine the eligibility of hematopoietic stem cell transplantation (HSCT).
Cancer genetic panel testing, which helps identifying mutations in 100-500 genes related to cancer, is increasingly used in clinic. In fact, two panel tests for solid tumors were approved by the PMDA (Pharmaceuticals and Medical Devices Agency of Japan) in June 2019; however, there is no such test available for HMs as of May 2020. Since 1) the spectrum of mutated genes is different from that seen in solid tumors and 2) implications of cancer genetic tests in clinical practice are distinct as discussed above, the development of a cancer genetic panel designed specifically for HMs is necessary. We have developed the DISCAVar (Diagnostic Sequencing of Cancer Associated Variants) panel which covers most mutations observed in HMs in collaboration with colleagues at the Department of Medicine and Biosystemic Science at Kyushu University School of Medicine and at the Center for Cellular and Molecular Medicine at Kyushu University Hospital. We are also developing an HM panel test, of which we plan to apply for a PMDA approval, in collaboration with scientists at National Cancer Center Japan, Kyushu University, Kyoto University, Nagoya Medical Center and Otsuka Pharmaceutical.
Cancer genetic panel testing is necessary but not sufficient to fully understand cancer. We will pursue clinical, translational and basic research to devise strategies to overcome cancer refractoriness.
The DISCAVar
(Diagnostic Sequencing of Cancer Associated Variants) panel
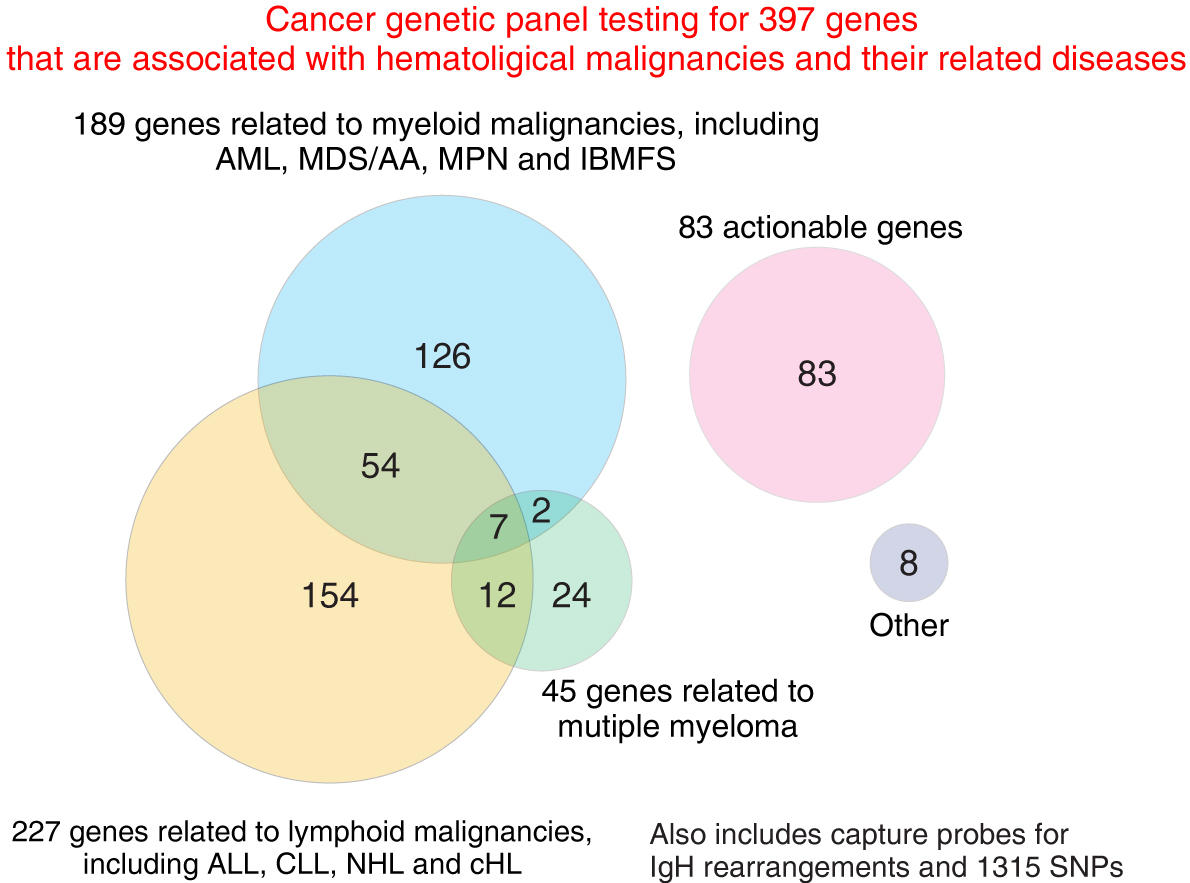
Figure 1 We have developed the DISCAVar (Diagnostic Sequencing of Cancer Associated Variants) cancer genetic panel, which covers most mutations observed in hematological malignancies and their related diseases, in collaboration with colleagues at the Department of Medicine and Biosystemic Science at Kyushu University School of Medicine and at the Center for Cellular and Molecular Medicine at Kyushu University Hospital. Semba et al. unpublished.
DISCAVar report
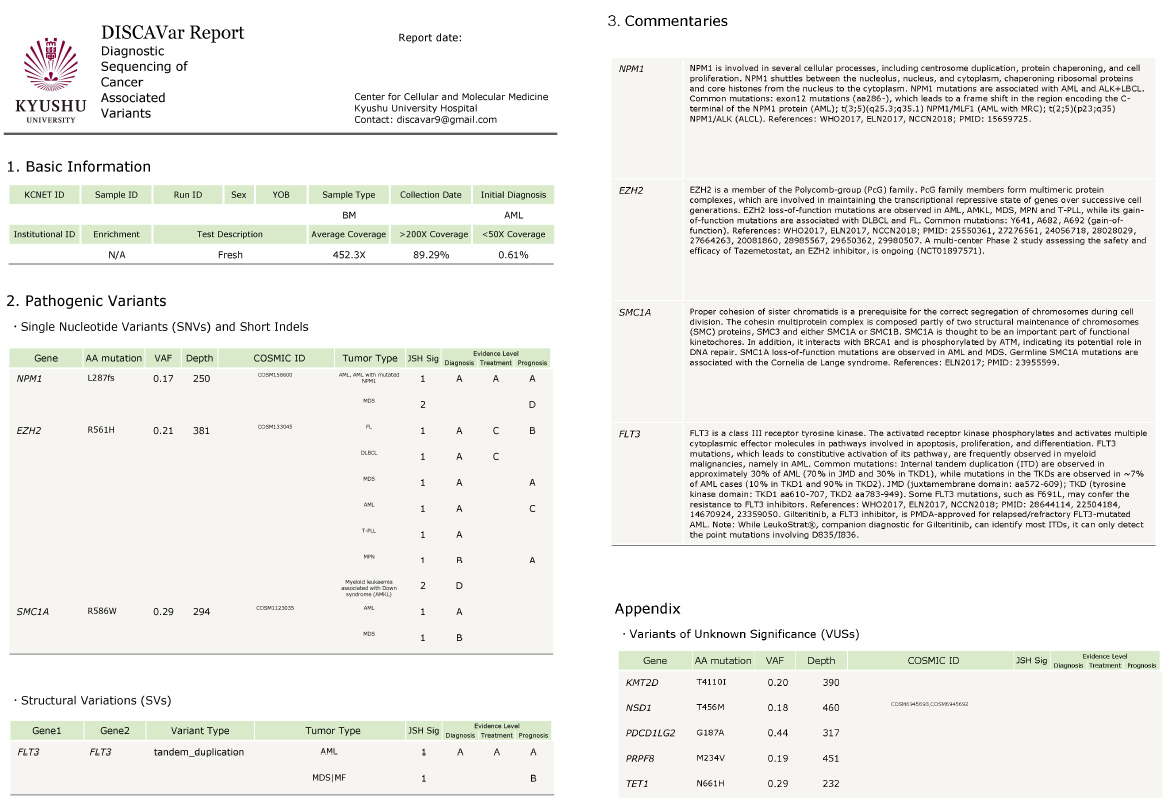
Figure 2 An example of the DISCAVar report. Sample information and QC data are described in the “Basic Information” section. Pathogenic variants and their VAFs (variant allele frequenciy), read depth and Cosmic IDs are listed in the “Pathogenic Variants” section. Clinical significance scores and evidence levels of each variant (defined by the Japanese Society of Hematology genomice panel testing guideline 2020) are also shown. Brief descriptions of mutated genes are described in the “Commentaries” section. Semba et al. unpublished.
Functional Cancer Genomics
Recent progress in sequencing technologies helps unveiling a near complete picture of the cancer genome. However, there are many unknowns as to 1) how mutated genes collaboratively function in cancer development and progression; 2) how gene mutations affect sensitivity and resistance to drug treatment and 3) whether cancer cells with a specific mutation exhibit synthetic lethal relationships with a specific drug treatment. We address these questions and develop novel strategies for cancer therapy employing CRISPR/Cas9 gene editing tools. We are particularly interested in identifying novel drug targets for acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL).
Genome-wide CRISPR-Cas9 screens to identify novel targets for AML therapy
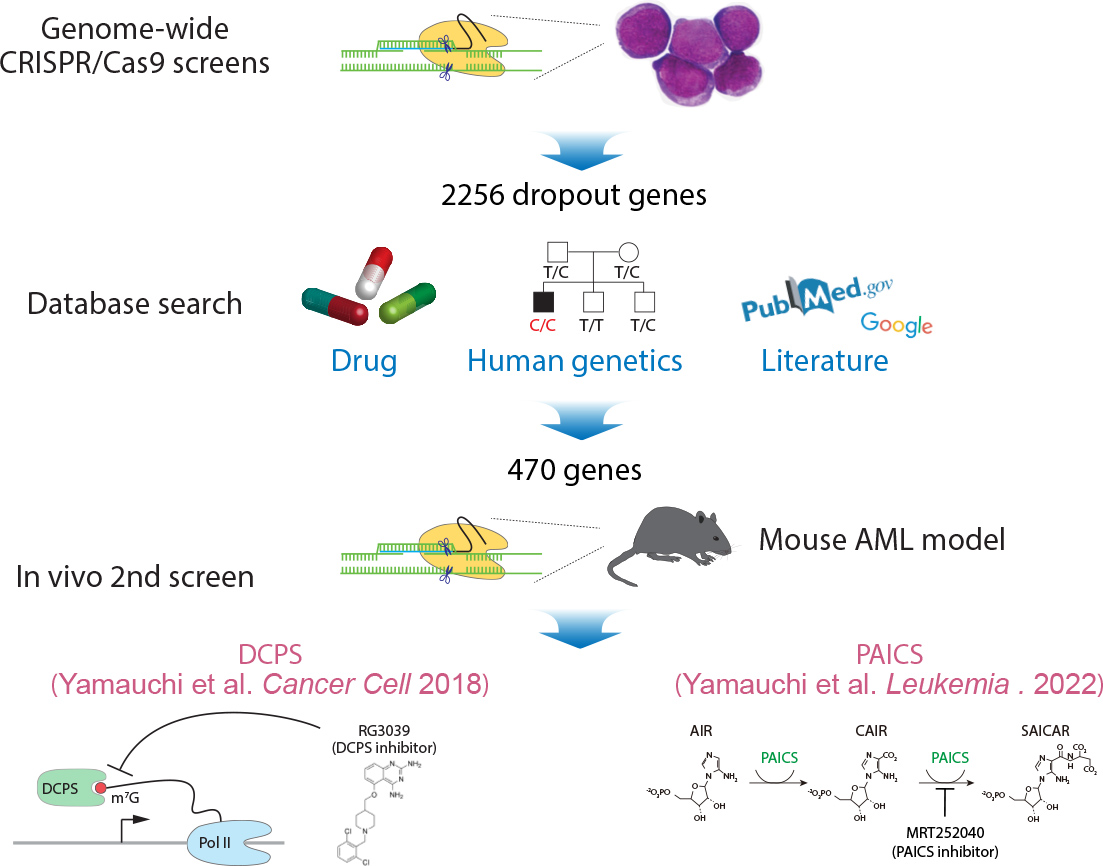
Figure 3 To identify novel targets for acute myeloid leukemia (AML) therapy, we employ genome-wide CRISPR-Cas9 screening using AML cell lines, followed by a second screen in vivo. We have identified DCPS (mRNA decapping enzyme scavenger) and PAICS (Phosphoribosylaminoimidazole carboxylase and SAICAR synthase) as novel therapeutic targets. Yamauchi et al. Cancer Cell 2018. Yamauchi et al. Leukemia . 2022
Our strategies: functional cancer genomics, followed by molecular analysis
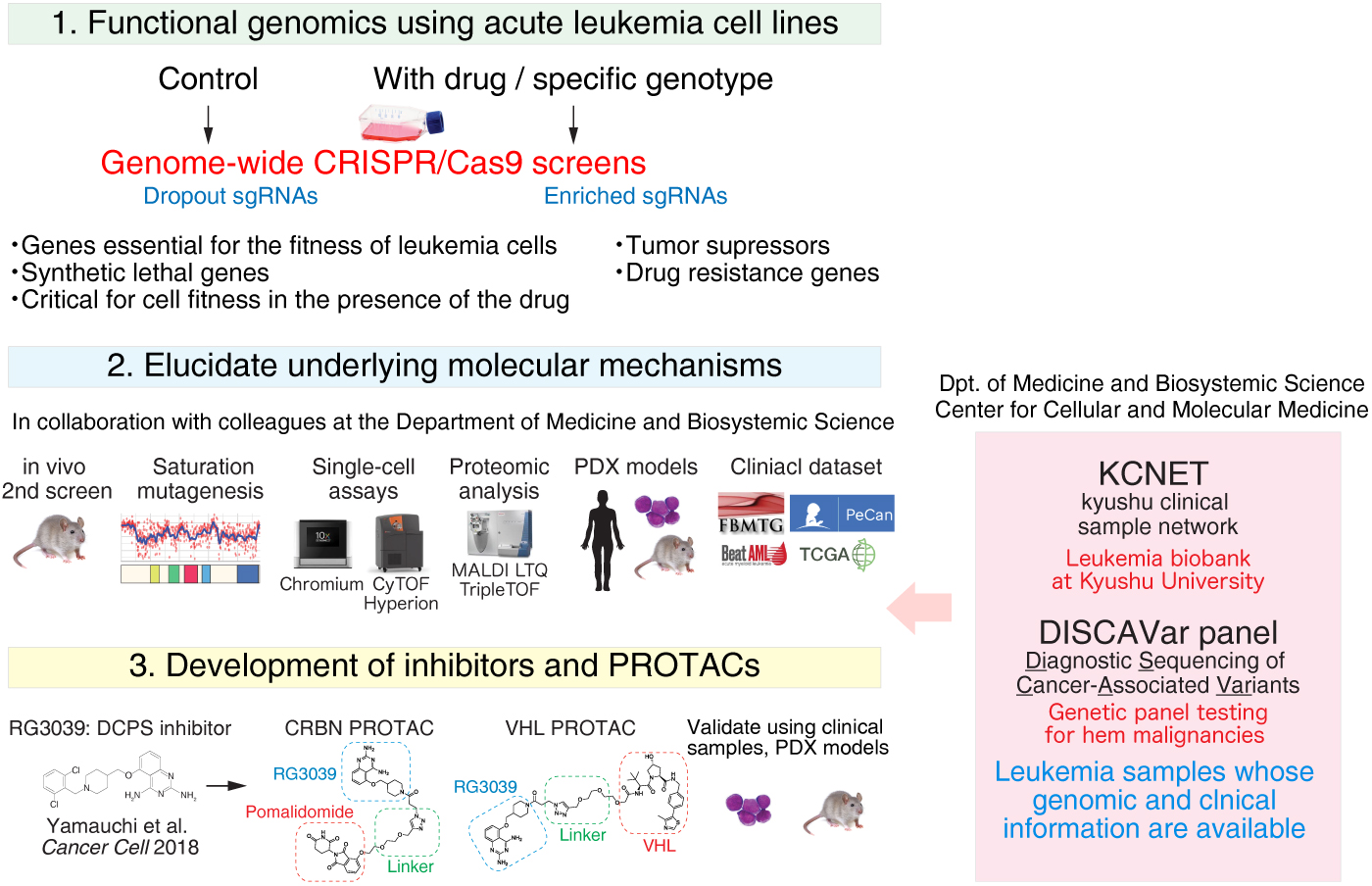
Figure 4 We perform genome-wide CRISPR/Cas9 screens in the presence or absence of a drug to identify novel drug resistant mechanisms and/or potential targets for combination therapies with that drug. We also generate a series of AML lines that lack a tumor supressor gene using the parental line and perform CRISPR screens to identify genes that exhibit synthetic lethal relationships with the tumor supressor. We validate screening results using molecular biological methods and elucidate molecular mechanisms underlying the obsereved phenotypes. Our long-term goal is to develop small molecules (e.g. inhibitors and PROTACs) for leukemia therapy in collaboration with our academic and industrial collaborators.
Single-cell Cancer Genomics
Intra- and inter clonal heterogeneity is a major cause for cancer recurrence. Since cancer cells in the body are heterogenous in their genome, epigenome, transcriptome and proteome, decoding them at the single-cell resolution is an intial step to elucidate clonal heterogeneity. To do so, we employ a series of single-cell technologies to decipher clonal heterogeneity of cancer.
Single-cell mRNA expression analysis of human bone marrow cells
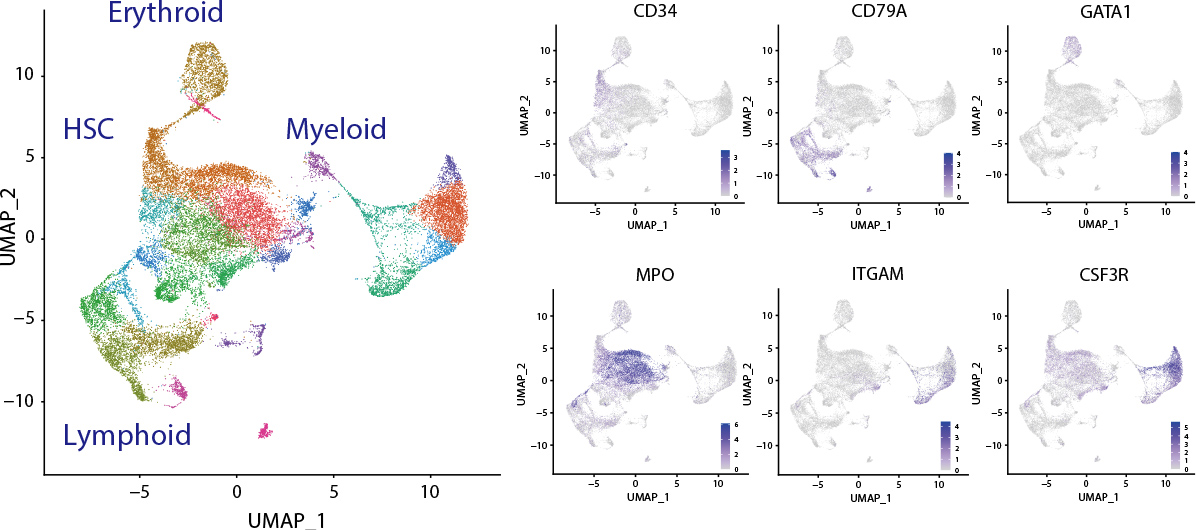
Figure 5 An example of single-cell transcriptomic analysis of human bone marrow cells. Lineage-positive mature fractions were removed prior to the analysis using magnetic beads. mRNA expression profiles of lineage-negative HSPCs (hematopoietic stem progenitor cells) were analyzed at single-cell level using the 10x Chromium system (left). Expression levels of lineage markers are also shown (right). Semba et al. unpublished.
Precision Hematology
Hematopoietic stem cells (HSCs) give rise to all hematopoietic lineage cells, including white blood cells (WBCs), which consists of myeloid and lymphoid cells, red blood cells (RBCs) and platelets. HSCs either self-renew or differentiate to hematopoietic progenitors, which differentiate and proliferate to give rise to mature cells in each lineage. Defects in this process due to a gene mutation(s) is a hallmark of hematologic malignancies (HMs): differentiation block and/or excessive proliferation generally underlie in the pathogenesis of HMs.
Hematopoietic stem cells (HSCs) give rise to all hematopoietic lineage cells

Figure 6 Hematopoietic stem cells (HSCs) self-renew and differentiate, giving rise to all hematopoietic lineadge cells. This figure shows hierarchy of hematopoietic development in mice.
We study molecular mechanisms underlying normal and malignant hematopoiesis “with precision” to devise novel strategies to treat HMs. We also study how fetal globin expression is silenced in adult erythroid cells aiming to find a means to pharmacologically induce fetal globin expression for the treatment of hemoglobinopathies.
Roles of the transcription factor LRF/ZBTB7A in hematopoiesis
There are 1,700-1,900 genes encoding transcription factors in the human genome, and at least 49 are classified as ZBTB proteins (broad-complex, tram-track and bric à brac-zinc finger transcription factors). This family plays critical roles in hematopoietic differentiation, lineage fate determination and malignant transformation. We have been interested in the roles of LRF (for leukemia lymphoma related factor, also known as ZBTB7A, FBI-1, OCZF and Pokemon) in normal and malignant hematopoiesis. Using conventional and conditional Zbtb7a knockout mouse models, we have identified key roles of LRF in HSC maintenance, early lymphoid fate decision, mature B cell development, germinal center formation and terminal erythroid differentiation.
Roles of the transcription factor LRF/ZBTB7A in the hematopoietic development
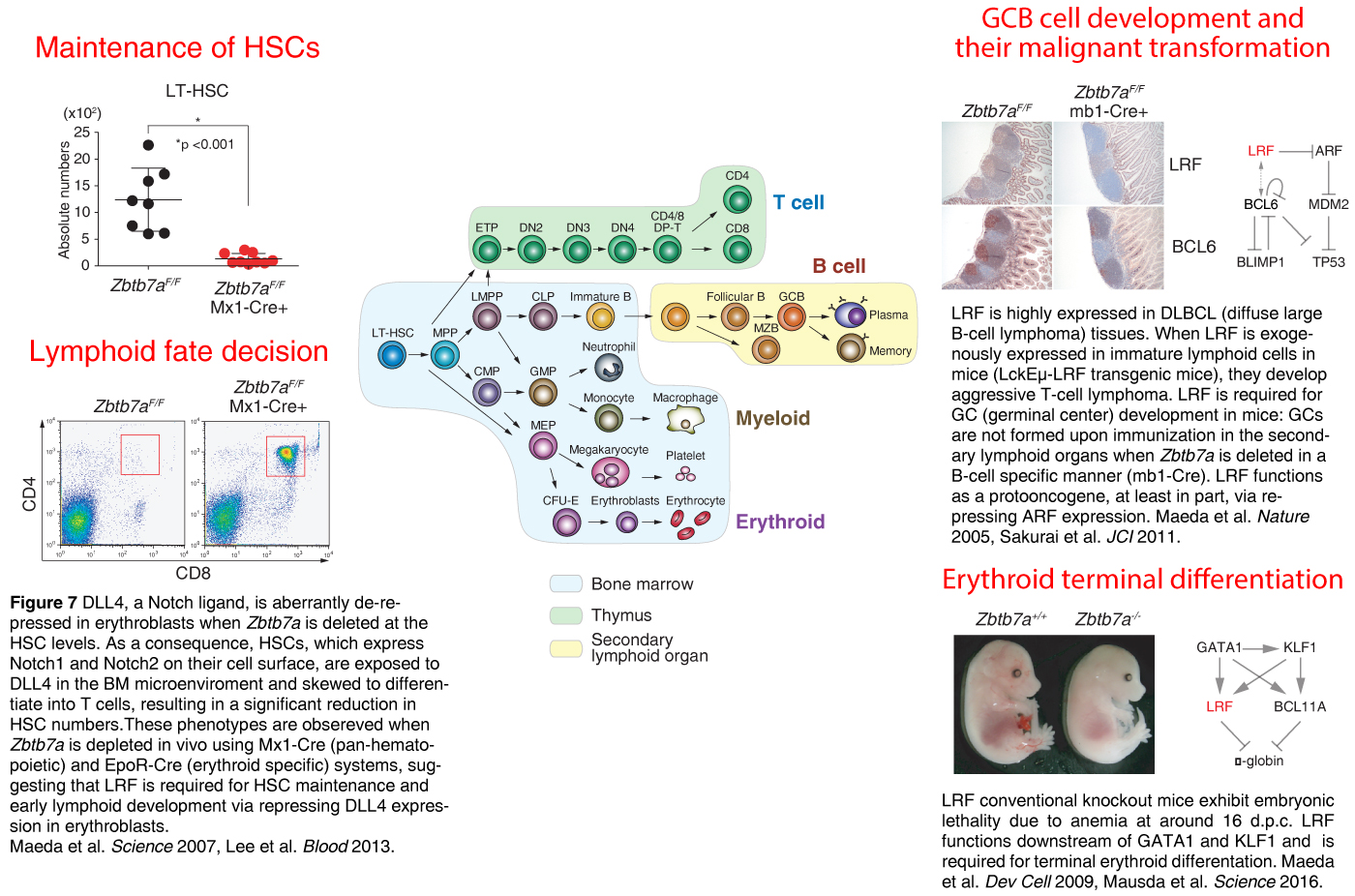
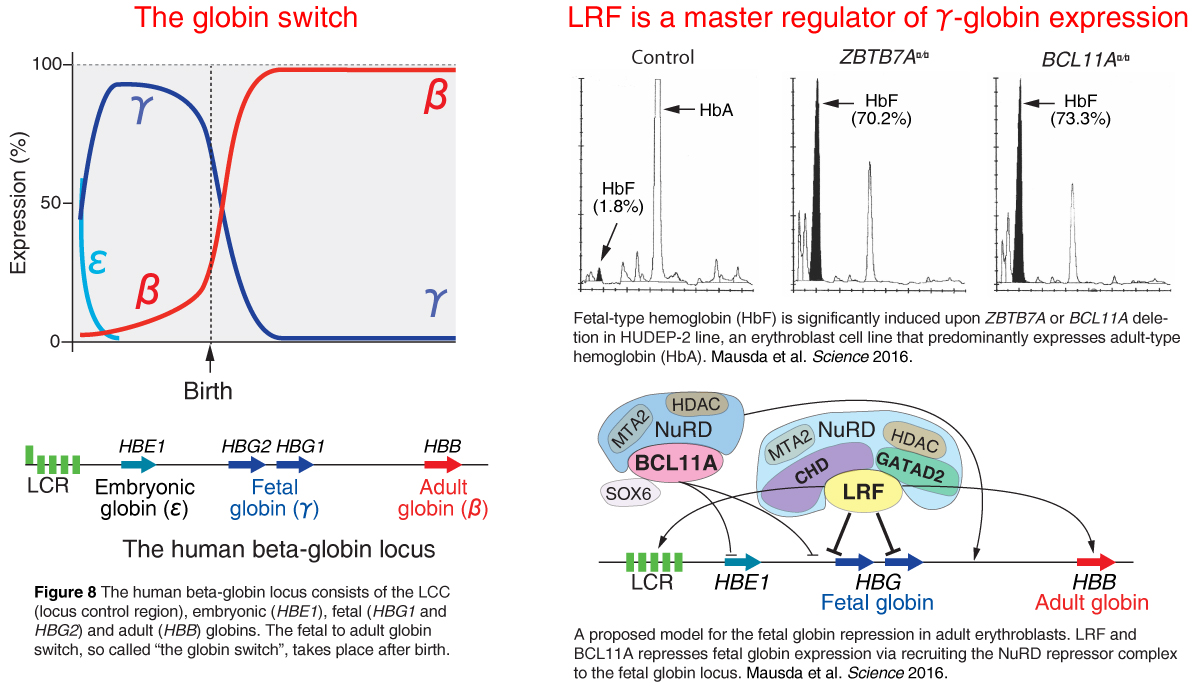
Mutations in adult globin genes cause hemoglobinopathies such as sickle cell disease (SCD) and thalassemia, which are among the most common monogenic inherited human disorders. Induction of fetal-type globin (γ-globin) is a promising means to treat these disorders; however, precisely how γ-globin expression is silenced in adult erythroid cells is not fully understood. We have uncovered a novel LRF function in the globin switch: LRF inactivation re-activates γ-globin expression in human adult erythroid cells independently of BCL11A, a master regulator of γ-globin expression. LRF presumably represses γ-globin expression through LRF-BTB/NuRD-mediated mechanisms.
LRF/ZBTB7A represses γ-globin expression in adult erythroblasts
PICALM is essential for transferrin receptor endocytosis in erythroblasts
PICALM, which encodes the clathrin adaptor protein PICALM, was originally identified as a component of the CALM-AF10 (PICALM-MLLT10) leukemia oncogene. To determine the role of Picalm function in adult hematopoiesis, we have generated a conditional knockout mouse strain in which Picalm can be deleted in a time- and tissue-specific manner. We have shown that PICALM is critical for clathrin-mediated transferrin endocytosis in erythroblasts; however, it was largely dispensable for myeloid and B-lymphoid development. While clathrin-dependent endocytosis has been thought to employ a “common machinery” shared by all cell types, our data suggest a cell type-specific endocytic machinery regulated by PICALM in erythroblasts. Furthermore, our study identifies molecular mechanisms required for efficient iron uptake in erythroblasts.
PICALM is essential for transferrin receptor (TFR) endocytosis in erythroblasts
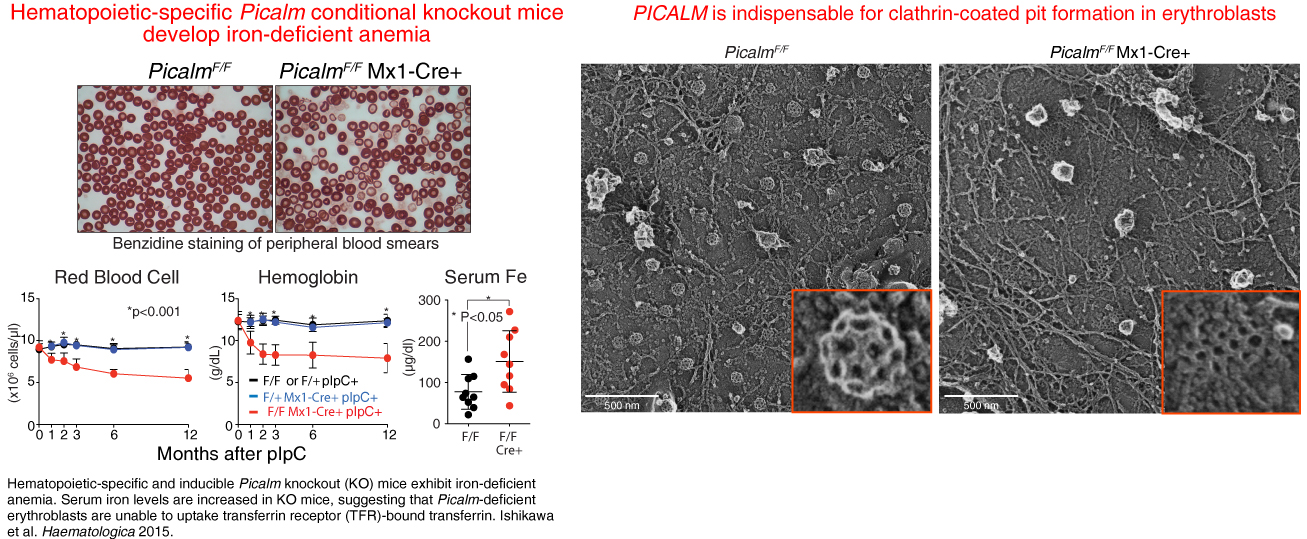
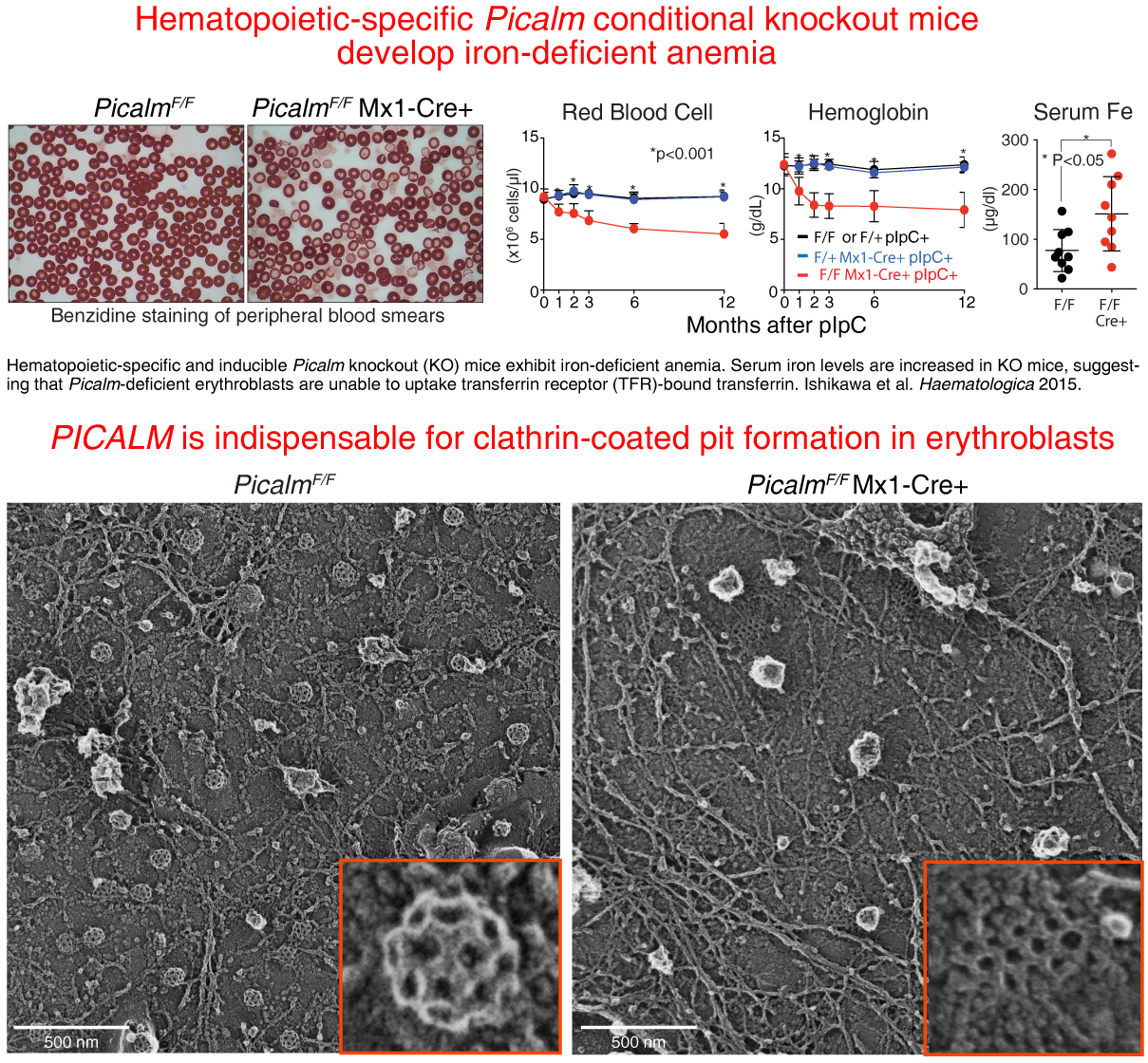
Figure 9 Freeze-etch electron microscopy (freeze-etch EM) images indicate a decrease in the number of fully-formed clathrin coated pits in Picalm-deficient erythroblasts (right) when compared to images from control cells (left). Ishikawa et al. Haematologica 2015.
CHIP (Clonal Hematopoiesis of Indeterminate Potential)
It has been thought that gene mutations are only seen in cancerous cells. However, recent studies suggest that HM-related gene mutations are also observed in the blood cells of old people. HSCs harboring such mutations presumably acquire a growth advantage over normal HSCs and dominate the hematopoietic system as people age. This phenomenon is called CHIP (Clonal Hematopoiesis of Indeterminate Potential). People with CHIP don’t exhibit symptoms of HMs or abnormal blood counts; however, their risk of developing HMs are nearly ten times higher than those without CHIP. Strikingly, individuals with CHIP also have an increased risk of developing cardiovascular disease. Since immune cells, whose dysfunction could cause inflammation, can migrate through circulation to every organ, it is possible that CHIP plays a broader role in the pathogenesis of age-related chronic inflammatory diseases. We are interested in elucidating the link between CHIP and chronic inflammatory diseases in collaboration with scientists from other fields.